
http://patrick.saintjean.free.fr/Teratec2020.html
http://patrick.saintjean.free.fr/
http://patrick.saintjean.free.fr/ChoixBibliographiquePSJ.html
Studio Virtuel Interactif en Réseau de Génie Biologique et Médical
sur le Génome en 3D interactif animatique
et simulation de processus biogénomiques et biogénétiques
avec Capteurs de Virus (pathologies et stratégies thérapeutiques).
Searching@Home à la Française pour permettre aux Chercheurs isolés
à former des Groupes de R&D locaux et internationaux à distance.
Studio Visio-Conférence interactive, inter-créative et inter-créactive.
Travaux en cours
MATH-LAB
A l'aide des Texturologies Quantiques, le High Parallel Computing (HPC) utilisant les Bits-Computers
devient un High Parallel Quantum Computing (HPQC) avec les QBits-Computers
et un Huge Parallel Texturology Quantum Computing (HUPTQC) avec les TQBits-Computers
et un Huge Parallel Optical Texturology Quantum Computing (HUPOTQC) avec les OTQBits-Computers (Obtical Texturology QBits)
Avec myQLM d'ATOS, nous préparons les NoteBooks myQLM de programmation quantique
The Atos Quantum Learning Machine (QLM & QLM E) is an enterprise-class solution
for quantum simulation that extends the capabilities of myQLM.
Avec Wolfram Mathematica les NoteBooks sont générés automatiquement à partir du calcul et de la programmation.
With the Jupyter notebooks, an open-source web application
that allows you to create and share documents that contain live code, equations, visualizations and narrative text
the data are cleaning for transformation, numerical simulation, statistical modeling, data visualization, machine learning and IA.
and With Binder, open those notebooks in an executable environment,
making your code immediately reproducible by anyone, anywhere.
https://gke.mybinder.org/
after enter your repository information by providing in the above form a URL or a GitHub repository that contains Jupyter notebooks,
as well as a branch, tag, or commit hash. Launch will build your Binder repository.
If you specify a path to a notebook file, the notebook will be opened in your browser after building.
A partir des travaux
sur la Trans-combinatoire et les Textures Prétopologiques et Quantiques et leurs Texturologies
ainsi que les Processus Markoviens Prétopologiques
associés à l'Algèbre des Quinternions comme système de trans-quaternions,
et à la Théorie des Sous-ensembles superposés et intriqués du Résualisme et de la Cybericité.
sont préparés avec Mathematica, des outils d'IA et DeepLearning, classification multi-hierarchique et multi-paramétrique,
avec représentation 2D et 3D interactives en temps réel par DataMining avec simulateur de vol dans les données en Réalité Virtuelle et Augmentée,
et algorithmique de Trans-Combinatoire (3^(n-1) + 1 possibilités en parallel au lieu de 2^n),
de Textures et Texturologies Quantiques Prétopologiques Relationnelles.
Ainsi est mise en place tout un processus de production d'outils et matériels,
de méthodes et démarches, d'interfaces de dialogue et de paramétrage en Design et UX-Design.
utilisables en Génie Biologique et Médical en Biochimie cellulaire et macrobiologie génômique.
Qu'est-ce que les modèles d'état de Markov?
Le pliage protéique est de nature
statistique, de sorte qu'une protéine peut se plier de plusieurs
façons. Nous avons besoin d'une carte pour pouvoir voir le tableau
d'ensemble.
Les modèles Markov State (MSM) sont une façon de décrire toutes les conformations (formes) qu'une protéine - ou d'autres biomolécules d'ailleurs - explore comme un ensemble d'états (c'est-à-dire des structures distinctes) et les taux de transition entre eux.
Ils établissent également les propriétés de mouvement et d’énergie de la protéine en se repliant d’une forme à l’autre. Une fois que nous avons toutes ces informations, nous pouvons observer les facteurs qui ont influencé le pliage, ce qui est particulièrement important si la protéine se déplie. Une grande partie de la théorie sous-jacente à ces méthodes est assez ancienne, mais leur utilisation a été limitée par les défis inhérents à l'identification d'un ensemble raisonnable d'États.
Les MSM nous sont particulièrement utiles car ils facilitent la parallélisation entre de nombreux processeurs informatiques en permettant l'agrégation statistique de courtes trajectoires de simulation indépendantes. Cela remplace la nécessité de trajectoires longues uniques et a donc été largement utilisé par les réseaux informatiques distribués tels que Folding@home et GPUGRID.
De plus, grâce à l'échantillonnage adaptatif, les MSM offrent un moyen d'accroître l'efficacité de la simulation sans introduire de biais ou d'approximations artificiels.
Beaucoup de progrès ont été fait en développant des méthodes de modèle d’état de Markov (MSM, Markov Stat Model, Model de Moor) pour analyser les données que nous produisons avec l’aide de la communauté F@H.
Plusieurs membres du Groupe Pande sont des Drs. Xuhui Huang et Gregory Bowman ont développé MSMBuilder, un logiciel open-source utilisé pour construire, analyser et visualiser les MSM.
Depuis sa sortie en 2009, il a été téléchargé plus de 1 600 fois sur les cinq continents et a été utilisé dans au moins 40 publications à ce jour.
Formellement, les MSM sont une application spécifique d'équations maîtres d'espace discret paramétrées à partir de la simulation.
Elles se composent de deux parties :
un système de partitionement de l'espace d'état X, généralement choisi pour diviser le système en un ensemble d'états métastables ;
et une équation principale décrivant la cinétique sur X, représentée par une matrice de transition T ou une matrice de vitesse.
L'espace d'état et l'équation de base sont tous deux trouvés à partir de la simulation moléculaire. La manière précise dont cela se fait varie considérablement.
What do MSMs look like?
Here are two examples:
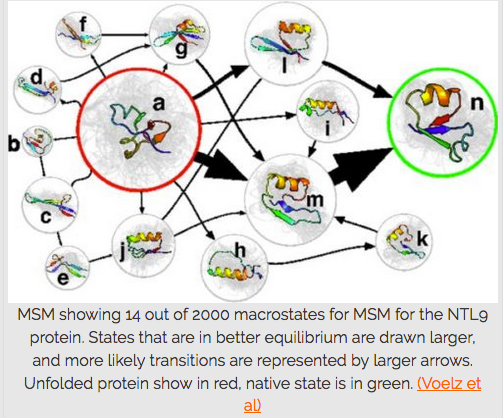

MSM montrant 14 macroétats sur 2000 pour les MSM de la protéine NTL9.
Les États qui sont en meilleur équilibre sont dessinés plus grands et les transitions les plus probables sont représentées par des flèches plus larges.
Les protéines dépliées sont en rouge, et l'état natif est en vert. (Voelz et al.)
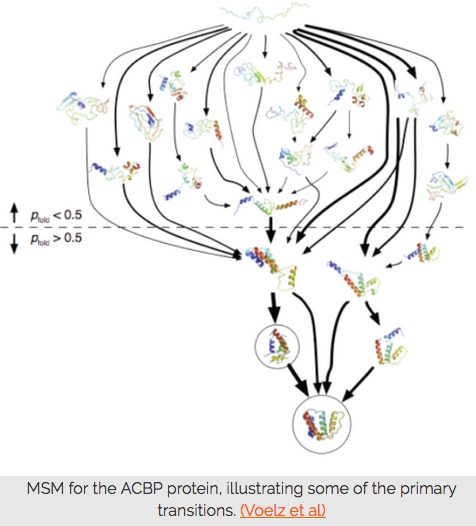
MSM pour la protéine ACBP, illustrant certaines des transitions primaires. (Voelz et al.)
Qu'est-ce que l'échantillonnage adaptatif, et comment est-il lié aux MSM ?
Lorsque les chercheurs utilisent des ordinateurs pour étudier la dynamique conformationnelle des protéines (la façon dont la protéine change de forme au fur et à mesure de son pliage), l’approche conventionnelle pour la dynamique moléculaire non biaisée de tous les atomes est en deux étapes. D'abord, ils exécutent un ensemble de simulations, et ensuite, une fois les simulations terminées, ils analysent les données obtenues.
L'approche adaptative de l'échantillonnage Markov State Model implique de rompre ce paradigme en entrelaçant ces deux étapes. Au lieu de construire le modèle uniquement après la collecte des données, il est construit à la volée au fur et à mesure que les données sont générées. Une boucle de rétroaction peut ensuite être mise en place lorsque l'état actuel du modèle est utilisé pour éclairer l'avancement de nouvelles simulations.
Imaginez, par exemple, que vous exploriez un labyrinthe pour la première fois. Bien que vous n’ayez pas de carte, vous avez un GPS qui vous permet de suivre vos progrès et d’afficher les parties du labyrinthe que vous avez exploré. Une approche est de mettre le GPS dans votre sac et de marcher aveuglément .. renverser les murs .. aussi longtemps que possible. Une fois fatigué, vous sortez le GPS et analysez le chemin que votre trajectoire a suivi ; en regardant votre chemin sur le GPS, vous pouvez voir la structure du labyrinthe et avoir effectivement construit une carte. Malheureusement, vous remarquez que vous avez perdu beaucoup de temps dans différentes parties du labyrinthe. Au lieu de cela, la stratégie la plus intelligente est de regarder le GPS en marchant... pour essayer de construire votre carte du labyrinthe progressivement. En utilisant votre carte, vous pouvez identifier quand vous êtes "coincé" dans une certaine partie du labyrinthe, et éviter de redécouvrir des parties du labyrinthe que vous êtes sûrs d’avoir déjà découvertes.
À bien des égards, ces deux approches de l'exploration d'un labyrinthe sont analogues aux deux approches de la collecte et de l'analyse de simulations moléculaires. En raison de la nature progressive de la construction du modèle à la volée dans l'approche d'échantillonnage adaptative, il est possible d'augmenter l'efficacité des simulations.
Comment créer un MSM à l'aide d'un échantillonnage adaptatif ?
Pour lancer un projet de simulation, nous devons d’abord choisir quelques conformations initiales (forme d’une protéine). Les méthodes heuristiques utilisées jusqu’à présent incluent l’exécution de simulations à haute température, l’utilisation de l’algorithme Monte Carlo de Rosetta et le choix asymétrique parmi les MSM apparentés de protéines similaires. (shooting off related MSMs of similar proteins)
Une fois un ensemble de conformations obtenu, chacune d'entre elles devient le point de départ de certaines simulations appelées ensemble un Run. A l'intérieur de chaque Course, de nombreuses trajectoires sont lancées, chacune appelée un Clone. Ainsi, tous les clones d'une course commencent à partir de la même forme protéique initiale, mais ils ont une vitesse initiale différente, c'est-à-dire que les atomes reçoivent une poussée initiale différente dans une direction ou une autre. Les clones d'une exécution peuvent trouver des conformations supplémentaires, auquel cas les extrémités de la série et plusieurs autres exécutions sont démarrées à partir d'elles.
Ce processus se poursuit avec beaucoup de Runs qui se ramifient à d'autres conformations, fusionnant peut-être ensemble à une forme commune avec d'autres Runs.
Au final, un modèle ayant des dizaines de milliers de conformations différentes, (téraoctets de données !) ayant :
- toutes les formes et les états d'énergie que la protéine peut prendre pendant son repliement vers son "état natif",
- les chances de toutes les transitions se produisant,
- et combien de temps il faut à la protéine pour terminer une transition d'une conformation à une autre.
Plus important encore, l'identification des endroits où les protéines se replient et se coincent, mène ensuite à plus de recherches et de modèles sur la façon d'empêcher cela de se produire.
Plus il y a d'ordinateurs participants, plus vite il est possible de compléter le Modèle d'État de Markov.
Qu’est-ce que les numéros PRCG ?
Les unités de travail sont étiquetées avec quatre numéros distincts dans le format :
Project (Run, Clone, Generation), Projet (Exécution, Clone, Génération).
Donc si le projet est la protéine à l'étude :
- un Run est une simulation lancée à partir d'une conformation particulière,
- et Runs contient de nombreux clones qui ont des vitesses initiales différentes.
Bien que Folding@home traite de nombreux projets, exécutions et clones différents en même temps, les clones eux-mêmes sont de nature série. Ils doivent être simulés du début à la fin, mais il serait peu pratique pour un ordinateur d'en terminer un seul. Au lieu de cela,
Votre ordinateur reçoit un morceau de clone.
La pièce est identifiée en utilisant le numéro Génération (Gen). Un ordinateur démarrera avec la Génération 0, et quand il finira, à un autre ordinateur sera donné la Génération 1, etc. Nous ne pouvons pas démarrer Gen 1 tant que le Gen 0 n'aura pas fini, et il peut y avoir des centaines de Gens. C'est pourquoi les unités de travail ont des délais et pourquoi la vitesse est si importante pour nous.
Pourquoi cette approche est-elle particulièrement utile ?
Cette approche peut être puissante car non seulement elle est très modifiable pour l'informatique distribuée, mais les ressources informatiques disponibles peuvent être utilisées plus efficacement.
Une protéine passe la plupart de son temps de pliage "coincée" dans une position énergétiquement favorable, avec des transitions - les processus en grande partie intéressants - n'ayant que rarement lieu.
De même, toute simulation simple du pliage de protéines perdra également du temps précieux à produire des données avec peu d'information.
Cependant, en utilisant le concept d'échantillonnage adaptatif, le modèle peut déterminer quand la simulation est bloquée,
puis réinitialiser de nouvelles simulations à partir de zones potentiellement plus fructueuses, en évitant le processus inutile de ré-exploration des zones déjà bien comprises.
Dans un article récent, les MSM ont été comparées à des méthodes de simulation plus traditionnelles comme les trajectoires de pliage très longues du superordinateur Anton, à un MSM construit à partir des mêmes données de pliage. Bien que notre MSM ait "découpé" la simulation en un tas de trajectoires courtes, il a été capable de reproduire très bien leurs simulations. De plus, l’approche des MSM a révélé de nouvelles idées sur le processus de pliage (une nouvelle voie de pliage) qui manquait dans l’approche plus traditionnelle d’ANTON.
Quelles sont les applications de ces techniques ?
Les MSM et l'échantillonnage adaptatif ont été utilisés pour étudier le pliage des protéines (1-8), la dynamique fonctionnelle (8-11), la liaison des ligands (11-14) et les interactions protéine-protéine (15).
Markov State Model (MSM) construction and validation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3462454/
The MSMBuilder33 software was used to build MSMs for ACBP under folding conditions (0M GuHCl, 330K simulations) and unfolding conditions (0.6–1.0 1M GuHCl, 370K simulations).
We found that a 20,000-microstate decomposition yielded a good balance of state connectivity and adequate transition sampling.
Conformations were clustered using a subset of 258 atoms (backbone N, Cα and C);
20% of the data was used to generate an initial clustering,
and the remaining 80% of the data was assigned to the generators.
The 20,000-microstate model was used for predicting experimental observables, while a 2000-macrostate MSM obtained by kinetic-based lumping 34 was used to analyze the distribution of folding pathway fluxes from unfolded to folded states.
Transition probabilities Tij of transitioning from state i to state j (within a lag time τ) are estimated by counting the number of transitions nij observed between time t and t+τ, and normalizing by rows:
Tij = nij/(Σj nij).
To enforce detailed balance, we symmetrize the forward and backward counts as (nij+nji)/(Σj nij+nji).
Artifacts from symmetrization are mostly limited to transitions with very few counts (and hence low populations that have negligible effects).
Sliding-window counts were used to alleviate finite-sampling errors.
To validate the robustness of these assumptions in estimating transition rates, we performed importance sampling of the posterior distribution of 2000-macrostate transition matrices, using a reversible conjugate prior for Markov chains as described in 35.
We generated ~5000 Markov chain realizations (samples of transition counts ñij, with no sliding window used; calculations are limited by storage space), from which expectation values (mean and variance) of equilibrium populations pi ∝ (Σj ñij) were calculated.
The expectation equilibrium populations calculated using the reversible prior were very similar to the symmetrization results (Supplementary Fig. S7e,f).
For example, the native macrostate population (pnat) using this procedure was 28.13% +/− 0.069%,
whereas the transition matrix constructed directly from from sliding-window counts yielded pnat = 30.3%, a discrepancy of only ~0.07 kT.
A lag time of τ=20 ns was determined to be suitable by building a series of MSMs at different lag times to find a region where the spectrum of implied timescales 36, 37
τi = −τ/ln(λi) are relatively insensitive to lag time.
To check the accuracy of the MSM, we compared average inter-residue distances over time (17–86, 1–86 and 17–50) seen in the trajectory data, to predictions from the MSM, and found reasonable agreement (see SI section B.1).
While the implied timescales become accelerated after lumping (it is difficult to achieve a perfect separation of timescales), distributions of folding pathway fluxes remain mostly intact for analysis.
A Bayesian inference model described in 38 was used to estimate Arrhenius barriers ΔG‡ij separating microstates and macrostates.
Committor (pfold) values and mean first passage times were computed for each macrostate using methods described in 37, 39.
The pfold values we compute for MSM macrostates are defined as the probability of reaching the native macrostate before the unfolded extended-chain macrostate.
Transition Path Theory (TPT) 40–42 was used to calculate pathways of reactive folding flux, using a modified “greedy backtracking” algorithm (see SI section B.2).
MSM equilibrium population vectors were calculated from the largest eigenvector of the transition matrix, i.e. from peq = peqT.
Macrostate free energies Fi were calculated from MSM equilibrium populations pi as Fi = −kT log pi at room temperature.
The free energy of folding as a function of the kinetic reaction coordinate pfold was calculated as F(pfold) = −kT log Z(pfold) where,
Z(pfold) = Σi χipi where χi is a bin indicator variable for bins with left edges
pfold = 0, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, 0.6, 0.7, 0.8, 0.9, 0.95.
Master Equation formalism
The continuous-time master equation describing the microstate dynamics is dp/dt = pK,
where p is the vector of state populations, and K is a 20,000 × 20,000 matrix of rate coefficients, related to the discrete-time transition probability matrix by T = exp(τK)
The solution of the master equation is
p(t) = ΣnψLn[ψRn · p(t = 0)] exp(λnt) = Σn pn(t),
where ψLn, ψRn, λn are the left and right eigenvectors and eigenvalues of K, respectively.
The kinetics can thus be described as a superposition of exponential relaxation modes pn(t) at implied timescales τ*n = −λn−1, each with amplitude an = [ψRn · p(t=0)].
MSM predictions of observables
Predicted values of observables over time were computed as F(t) = p(t) · f, where p(t) is a vector of state populations over time, and f is a vector of observables values for each microstate.
Uncertainty estimates were propagated assuming statistical independence of each state.
For some observables, time courses were obtained by discrete propagation of the transition probability matrix T, using p(t+τ) = p(t)T.
For others, p(t) was calculated from the 1000 slowest relaxation modes of the master equation solution.
RMSD pseudo-trajectories were calculated using a simple Monte Carlo algorithm to generate a trajectory of (20 ns) microstate jumps,
and selecting at random (uniformly) a simulation snapshot to report observables at each time step (see SI section B.3 for more examples).
Predictions of FRET observables over time were computed with special corrections for FRET probe linkers not present in the simulations (see SI section B.4),
and corrections for native state stability (see below).
Trp-Cys quenching rates and intramolecular diffusion coefficients for T17C-W58 and W58-I86C were predicted using methods described in 25
from simulated distributions of intramolecular Trp-Cys distances P(r) calculated from simulated unfolded ensembles (330 K, 0 M GuHCl and 370 K, 0.6–1.0 M GuHCl, starting from extended
and coil states, snapshots taken after 1 µs), where r is the distance between side-chain centroids (see also SI section A.5).
Intramolecular diffusion coefficients D were computed from trajectory data, by fitting the mean-squared displacements of Trp-Cys distances over time in blocks of 50 ns (sampled in 1-ns intervals),
as described previously.
Correcting predicted FRET values for native-state stability
A consequence of symmetrization of the transition probability matrix is that the equilibrium populations are proportional to the total number of observed counts : pi ∝ (Σj nij).
Because of this, the MSM predicts an equilibrium distribution of states with ~2:1 unfolded vs. folded populations, even under folding conditions.
To correct predicted observables, we compute FRET values by subtracting the equilibrium unfolded-state component of the signal (i.e. we assume that the simulated unfolded state is “invisible”).
The stationary state peq = (ncoil + next + nnat)/(Ncoil + Next + Nnat) is the (normalized) number of counts observed in the trajectories, where ncoil, next, and nnat are the vectors of observed microstate counts for simulations initiated from coil, extended and native states, respectively, and N = Ncoil + Next + Nnat is the total number of counts observed in all simulations.
We propagate the discrete-time transition matrix as described above to get populations over time, and calculate FRET using a modified projection operator S′:
S'(p) = (N/Nnat) · [S(p)−S([next + ncoil]/N)]
This projection operator has the property that as t→∞, S'(p(t→∞)) = S(nnat/Nnat). We use this correction for the FRET predictions in Figure 2d, setting the starting configuration p(t=0) to a single microstate corresponding to the extended state. A caveat of this approach is that negative FRET values may be obtained at very early times, when initial popultions are from unfolded states. For all cases we considered, we find that this effect only occurs for t < 1 µs, faster than the time resolution of the mixer experiments with which we make comparisons.
Les modèles Markov State (MSM) sont une façon de décrire toutes les conformations (formes) qu'une protéine - ou d'autres biomolécules d'ailleurs - explore comme un ensemble d'états (c'est-à-dire des structures distinctes) et les taux de transition entre eux.
Ils établissent également les propriétés de mouvement et d’énergie de la protéine en se repliant d’une forme à l’autre. Une fois que nous avons toutes ces informations, nous pouvons observer les facteurs qui ont influencé le pliage, ce qui est particulièrement important si la protéine se déplie. Une grande partie de la théorie sous-jacente à ces méthodes est assez ancienne, mais leur utilisation a été limitée par les défis inhérents à l'identification d'un ensemble raisonnable d'États.
Les MSM nous sont particulièrement utiles car ils facilitent la parallélisation entre de nombreux processeurs informatiques en permettant l'agrégation statistique de courtes trajectoires de simulation indépendantes. Cela remplace la nécessité de trajectoires longues uniques et a donc été largement utilisé par les réseaux informatiques distribués tels que Folding@home et GPUGRID.
De plus, grâce à l'échantillonnage adaptatif, les MSM offrent un moyen d'accroître l'efficacité de la simulation sans introduire de biais ou d'approximations artificiels.
Beaucoup de progrès ont été fait en développant des méthodes de modèle d’état de Markov (MSM, Markov Stat Model, Model de Moor) pour analyser les données que nous produisons avec l’aide de la communauté F@H.
Plusieurs membres du Groupe Pande sont des Drs. Xuhui Huang et Gregory Bowman ont développé MSMBuilder, un logiciel open-source utilisé pour construire, analyser et visualiser les MSM.
Depuis sa sortie en 2009, il a été téléchargé plus de 1 600 fois sur les cinq continents et a été utilisé dans au moins 40 publications à ce jour.
Formellement, les MSM sont une application spécifique d'équations maîtres d'espace discret paramétrées à partir de la simulation.
Elles se composent de deux parties :
un système de partitionement de l'espace d'état X, généralement choisi pour diviser le système en un ensemble d'états métastables ;
et une équation principale décrivant la cinétique sur X, représentée par une matrice de transition T ou une matrice de vitesse.
L'espace d'état et l'équation de base sont tous deux trouvés à partir de la simulation moléculaire. La manière précise dont cela se fait varie considérablement.
What do MSMs look like?
Here are two examples:
MSM montrant 14 macroétats sur 2000 pour les MSM de la protéine NTL9.
Les États qui sont en meilleur équilibre sont dessinés plus grands et les transitions les plus probables sont représentées par des flèches plus larges.
Les protéines dépliées sont en rouge, et l'état natif est en vert. (Voelz et al.)
Qu'est-ce que l'échantillonnage adaptatif, et comment est-il lié aux MSM ?
Lorsque les chercheurs utilisent des ordinateurs pour étudier la dynamique conformationnelle des protéines (la façon dont la protéine change de forme au fur et à mesure de son pliage), l’approche conventionnelle pour la dynamique moléculaire non biaisée de tous les atomes est en deux étapes. D'abord, ils exécutent un ensemble de simulations, et ensuite, une fois les simulations terminées, ils analysent les données obtenues.
L'approche adaptative de l'échantillonnage Markov State Model implique de rompre ce paradigme en entrelaçant ces deux étapes. Au lieu de construire le modèle uniquement après la collecte des données, il est construit à la volée au fur et à mesure que les données sont générées. Une boucle de rétroaction peut ensuite être mise en place lorsque l'état actuel du modèle est utilisé pour éclairer l'avancement de nouvelles simulations.
Imaginez, par exemple, que vous exploriez un labyrinthe pour la première fois. Bien que vous n’ayez pas de carte, vous avez un GPS qui vous permet de suivre vos progrès et d’afficher les parties du labyrinthe que vous avez exploré. Une approche est de mettre le GPS dans votre sac et de marcher aveuglément .. renverser les murs .. aussi longtemps que possible. Une fois fatigué, vous sortez le GPS et analysez le chemin que votre trajectoire a suivi ; en regardant votre chemin sur le GPS, vous pouvez voir la structure du labyrinthe et avoir effectivement construit une carte. Malheureusement, vous remarquez que vous avez perdu beaucoup de temps dans différentes parties du labyrinthe. Au lieu de cela, la stratégie la plus intelligente est de regarder le GPS en marchant... pour essayer de construire votre carte du labyrinthe progressivement. En utilisant votre carte, vous pouvez identifier quand vous êtes "coincé" dans une certaine partie du labyrinthe, et éviter de redécouvrir des parties du labyrinthe que vous êtes sûrs d’avoir déjà découvertes.
À bien des égards, ces deux approches de l'exploration d'un labyrinthe sont analogues aux deux approches de la collecte et de l'analyse de simulations moléculaires. En raison de la nature progressive de la construction du modèle à la volée dans l'approche d'échantillonnage adaptative, il est possible d'augmenter l'efficacité des simulations.
Comment créer un MSM à l'aide d'un échantillonnage adaptatif ?
Pour lancer un projet de simulation, nous devons d’abord choisir quelques conformations initiales (forme d’une protéine). Les méthodes heuristiques utilisées jusqu’à présent incluent l’exécution de simulations à haute température, l’utilisation de l’algorithme Monte Carlo de Rosetta et le choix asymétrique parmi les MSM apparentés de protéines similaires. (shooting off related MSMs of similar proteins)
Une fois un ensemble de conformations obtenu, chacune d'entre elles devient le point de départ de certaines simulations appelées ensemble un Run. A l'intérieur de chaque Course, de nombreuses trajectoires sont lancées, chacune appelée un Clone. Ainsi, tous les clones d'une course commencent à partir de la même forme protéique initiale, mais ils ont une vitesse initiale différente, c'est-à-dire que les atomes reçoivent une poussée initiale différente dans une direction ou une autre. Les clones d'une exécution peuvent trouver des conformations supplémentaires, auquel cas les extrémités de la série et plusieurs autres exécutions sont démarrées à partir d'elles.
Ce processus se poursuit avec beaucoup de Runs qui se ramifient à d'autres conformations, fusionnant peut-être ensemble à une forme commune avec d'autres Runs.
Au final, un modèle ayant des dizaines de milliers de conformations différentes, (téraoctets de données !) ayant :
- toutes les formes et les états d'énergie que la protéine peut prendre pendant son repliement vers son "état natif",
- les chances de toutes les transitions se produisant,
- et combien de temps il faut à la protéine pour terminer une transition d'une conformation à une autre.
Plus important encore, l'identification des endroits où les protéines se replient et se coincent, mène ensuite à plus de recherches et de modèles sur la façon d'empêcher cela de se produire.
Plus il y a d'ordinateurs participants, plus vite il est possible de compléter le Modèle d'État de Markov.
Qu’est-ce que les numéros PRCG ?
Les unités de travail sont étiquetées avec quatre numéros distincts dans le format :
Project (Run, Clone, Generation), Projet (Exécution, Clone, Génération).
Donc si le projet est la protéine à l'étude :
- un Run est une simulation lancée à partir d'une conformation particulière,
- et Runs contient de nombreux clones qui ont des vitesses initiales différentes.
Bien que Folding@home traite de nombreux projets, exécutions et clones différents en même temps, les clones eux-mêmes sont de nature série. Ils doivent être simulés du début à la fin, mais il serait peu pratique pour un ordinateur d'en terminer un seul. Au lieu de cela,
Votre ordinateur reçoit un morceau de clone.
La pièce est identifiée en utilisant le numéro Génération (Gen). Un ordinateur démarrera avec la Génération 0, et quand il finira, à un autre ordinateur sera donné la Génération 1, etc. Nous ne pouvons pas démarrer Gen 1 tant que le Gen 0 n'aura pas fini, et il peut y avoir des centaines de Gens. C'est pourquoi les unités de travail ont des délais et pourquoi la vitesse est si importante pour nous.
Pourquoi cette approche est-elle particulièrement utile ?
Cette approche peut être puissante car non seulement elle est très modifiable pour l'informatique distribuée, mais les ressources informatiques disponibles peuvent être utilisées plus efficacement.
Une protéine passe la plupart de son temps de pliage "coincée" dans une position énergétiquement favorable, avec des transitions - les processus en grande partie intéressants - n'ayant que rarement lieu.
De même, toute simulation simple du pliage de protéines perdra également du temps précieux à produire des données avec peu d'information.
Cependant, en utilisant le concept d'échantillonnage adaptatif, le modèle peut déterminer quand la simulation est bloquée,
puis réinitialiser de nouvelles simulations à partir de zones potentiellement plus fructueuses, en évitant le processus inutile de ré-exploration des zones déjà bien comprises.
Dans un article récent, les MSM ont été comparées à des méthodes de simulation plus traditionnelles comme les trajectoires de pliage très longues du superordinateur Anton, à un MSM construit à partir des mêmes données de pliage. Bien que notre MSM ait "découpé" la simulation en un tas de trajectoires courtes, il a été capable de reproduire très bien leurs simulations. De plus, l’approche des MSM a révélé de nouvelles idées sur le processus de pliage (une nouvelle voie de pliage) qui manquait dans l’approche plus traditionnelle d’ANTON.
Quelles sont les applications de ces techniques ?
Les MSM et l'échantillonnage adaptatif ont été utilisés pour étudier le pliage des protéines (1-8), la dynamique fonctionnelle (8-11), la liaison des ligands (11-14) et les interactions protéine-protéine (15).
Markov State Model (MSM) construction and validation
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3462454/
The MSMBuilder33 software was used to build MSMs for ACBP under folding conditions (0M GuHCl, 330K simulations) and unfolding conditions (0.6–1.0 1M GuHCl, 370K simulations).
We found that a 20,000-microstate decomposition yielded a good balance of state connectivity and adequate transition sampling.
Conformations were clustered using a subset of 258 atoms (backbone N, Cα and C);
20% of the data was used to generate an initial clustering,
and the remaining 80% of the data was assigned to the generators.
The 20,000-microstate model was used for predicting experimental observables, while a 2000-macrostate MSM obtained by kinetic-based lumping 34 was used to analyze the distribution of folding pathway fluxes from unfolded to folded states.
Transition probabilities Tij of transitioning from state i to state j (within a lag time τ) are estimated by counting the number of transitions nij observed between time t and t+τ, and normalizing by rows:
Tij = nij/(Σj nij).
To enforce detailed balance, we symmetrize the forward and backward counts as (nij+nji)/(Σj nij+nji).
Artifacts from symmetrization are mostly limited to transitions with very few counts (and hence low populations that have negligible effects).
Sliding-window counts were used to alleviate finite-sampling errors.
To validate the robustness of these assumptions in estimating transition rates, we performed importance sampling of the posterior distribution of 2000-macrostate transition matrices, using a reversible conjugate prior for Markov chains as described in 35.
We generated ~5000 Markov chain realizations (samples of transition counts ñij, with no sliding window used; calculations are limited by storage space), from which expectation values (mean and variance) of equilibrium populations pi ∝ (Σj ñij) were calculated.
The expectation equilibrium populations calculated using the reversible prior were very similar to the symmetrization results (Supplementary Fig. S7e,f).
For example, the native macrostate population (pnat) using this procedure was 28.13% +/− 0.069%,
whereas the transition matrix constructed directly from from sliding-window counts yielded pnat = 30.3%, a discrepancy of only ~0.07 kT.
A lag time of τ=20 ns was determined to be suitable by building a series of MSMs at different lag times to find a region where the spectrum of implied timescales 36, 37
τi = −τ/ln(λi) are relatively insensitive to lag time.
To check the accuracy of the MSM, we compared average inter-residue distances over time (17–86, 1–86 and 17–50) seen in the trajectory data, to predictions from the MSM, and found reasonable agreement (see SI section B.1).
While the implied timescales become accelerated after lumping (it is difficult to achieve a perfect separation of timescales), distributions of folding pathway fluxes remain mostly intact for analysis.
A Bayesian inference model described in 38 was used to estimate Arrhenius barriers ΔG‡ij separating microstates and macrostates.
Committor (pfold) values and mean first passage times were computed for each macrostate using methods described in 37, 39.
The pfold values we compute for MSM macrostates are defined as the probability of reaching the native macrostate before the unfolded extended-chain macrostate.
Transition Path Theory (TPT) 40–42 was used to calculate pathways of reactive folding flux, using a modified “greedy backtracking” algorithm (see SI section B.2).
MSM equilibrium population vectors were calculated from the largest eigenvector of the transition matrix, i.e. from peq = peqT.
Macrostate free energies Fi were calculated from MSM equilibrium populations pi as Fi = −kT log pi at room temperature.
The free energy of folding as a function of the kinetic reaction coordinate pfold was calculated as F(pfold) = −kT log Z(pfold) where,
Z(pfold) = Σi χipi where χi is a bin indicator variable for bins with left edges
pfold = 0, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, 0.6, 0.7, 0.8, 0.9, 0.95.
Master Equation formalism
The continuous-time master equation describing the microstate dynamics is dp/dt = pK,
where p is the vector of state populations, and K is a 20,000 × 20,000 matrix of rate coefficients, related to the discrete-time transition probability matrix by T = exp(τK)
The solution of the master equation is
p(t) = ΣnψLn[ψRn · p(t = 0)] exp(λnt) = Σn pn(t),
where ψLn, ψRn, λn are the left and right eigenvectors and eigenvalues of K, respectively.
The kinetics can thus be described as a superposition of exponential relaxation modes pn(t) at implied timescales τ*n = −λn−1, each with amplitude an = [ψRn · p(t=0)].
MSM predictions of observables
Predicted values of observables over time were computed as F(t) = p(t) · f, where p(t) is a vector of state populations over time, and f is a vector of observables values for each microstate.
Uncertainty estimates were propagated assuming statistical independence of each state.
For some observables, time courses were obtained by discrete propagation of the transition probability matrix T, using p(t+τ) = p(t)T.
For others, p(t) was calculated from the 1000 slowest relaxation modes of the master equation solution.
RMSD pseudo-trajectories were calculated using a simple Monte Carlo algorithm to generate a trajectory of (20 ns) microstate jumps,
and selecting at random (uniformly) a simulation snapshot to report observables at each time step (see SI section B.3 for more examples).
Predictions of FRET observables over time were computed with special corrections for FRET probe linkers not present in the simulations (see SI section B.4),
and corrections for native state stability (see below).
Trp-Cys quenching rates and intramolecular diffusion coefficients for T17C-W58 and W58-I86C were predicted using methods described in 25
from simulated distributions of intramolecular Trp-Cys distances P(r) calculated from simulated unfolded ensembles (330 K, 0 M GuHCl and 370 K, 0.6–1.0 M GuHCl, starting from extended
and coil states, snapshots taken after 1 µs), where r is the distance between side-chain centroids (see also SI section A.5).
Intramolecular diffusion coefficients D were computed from trajectory data, by fitting the mean-squared displacements of Trp-Cys distances over time in blocks of 50 ns (sampled in 1-ns intervals),
as described previously.
Correcting predicted FRET values for native-state stability
A consequence of symmetrization of the transition probability matrix is that the equilibrium populations are proportional to the total number of observed counts : pi ∝ (Σj nij).
Because of this, the MSM predicts an equilibrium distribution of states with ~2:1 unfolded vs. folded populations, even under folding conditions.
To correct predicted observables, we compute FRET values by subtracting the equilibrium unfolded-state component of the signal (i.e. we assume that the simulated unfolded state is “invisible”).
The stationary state peq = (ncoil + next + nnat)/(Ncoil + Next + Nnat) is the (normalized) number of counts observed in the trajectories, where ncoil, next, and nnat are the vectors of observed microstate counts for simulations initiated from coil, extended and native states, respectively, and N = Ncoil + Next + Nnat is the total number of counts observed in all simulations.
We propagate the discrete-time transition matrix as described above to get populations over time, and calculate FRET using a modified projection operator S′:
S'(p) = (N/Nnat) · [S(p)−S([next + ncoil]/N)]
This projection operator has the property that as t→∞, S'(p(t→∞)) = S(nnat/Nnat). We use this correction for the FRET predictions in Figure 2d, setting the starting configuration p(t=0) to a single microstate corresponding to the extended state. A caveat of this approach is that negative FRET values may be obtained at very early times, when initial popultions are from unfolded states. For all cases we considered, we find that this effect only occurs for t < 1 µs, faster than the time resolution of the mixer experiments with which we make comparisons.